
Translate this page into:
An Indian infant with de novo duplication of 16p chromosome: A rare genetic syndrome
*Corresponding author: Manisha Goyal, Fellow Medical Genetics, Center of Rare Disease, Department of Pediatrics, J. K. Lon Hospital, SMS Medical College and Hospital, Jaipur, Rajasthan, India. manidr2000@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Goyal M, Gupta A, Faruq M, Shrivastava D. An Indian infant with de novo duplication of 16p chromosome: A rare genetic syndrome. Indian J Med Sci 2021;73(2):266-8.
Abstract
Facial dysmorphism along with multiple congenital anomalies is observed in many genetic syndromes mostly in chromosomal microdeletion or duplication, which cannot be detected by conventional karyotype. Here, we report a case with facial dysmorphism, cleft palate, congenital heart defect, and umbilical hernia, diagnosed with duplication at chromosome 16p13.3 by array comparative genomic hybridization (CGH) at very early age. Array CGH is the advanced diagnostic technology; enable to diagnose chromosomal abnormalities earlier thus can provide appropriate medical management and prognostication.
Keywords
Array comparative genomic hybridization
16 p duplication
Facial dysmorphism
Microdeletion

INTRODUCTION
High resolution array comparative genomic hybridization (array CGH) has proven a powerful tool to detect submicroscopic chromosomal imbalance associated with development delay/ intellectual disability with or without facial dysmorphism. Low copy repeats (LCRs) mediated non-allelic homologous recombination leading to the occurrence of microdeletion/duplication syndrome.[1] Short arm of chromosomal 16 (16p) is compassed of highly complex LCRs. 16p duplication syndrome is characterized by developmental delay, intellectual disability, growth and mental deficiency, facial dysmorphism, congenital heart, and urogenital anomalies.[2] The most critical region is 16 p13.1–13.3 in determining the clinical manifestation.[3,4]
We hereby report a 2 months old girl with 6.1 Mb gain on Chromosome 16p13.3 by array CGH, presented with growth retardation, facial dysmorphism, cleft palate, umbilical hernia, congenital heart defect, seizures, and spasticity of the lower limb. Early detection and discovery of phenotype-genotype associations broadens the understanding of development of affected child and prognosticate the outcome.
CASE REPORT
A 2-month-old girl was referred for evaluation of facial dysmorphism. The child was first born of non-consanguineous parents with uneventful antenatal and perinatal period. She was born at term by normal vaginal delivery with birth weight of 2.4 kg. She was admitted on day 3 with excessive cry and abnormal movements of body, detected with hypocalcemic seizures. Anthropometric parameters at 2 months of age were weight 3.4 kg (<3rd centile), height 52cm (15th–25th centile), and head circumference 35 cm (<3 centile). Facial features suggested open anterior fontanel, hypertelorism, sparse eyebrows and eyelashes, broad nasal bridge and tip, bilateral ptosis, bilateral low set cup shape ears, long and prominent philtrum, downturned corner of mouth, micrognathia, and anterior cleft palate [Figure 1]. There were bilateral simian crease and clinodactyly. Small umbilical hernia was present. Her tone in lower limb was increased and reflexes were present. Echocardiography suggested atrial septal defect (ASD). Ultrasonography abdomen and MRI brain were normal. Chromosomal analysis by karyotype and FISH analysis for 22q11.2 microdeletion was normal. Blood sample was sent for array CGH in EDTA vial. Cytogenetic study was conducted using Affymetrix CytoScan 750 K array (Affymetrix Inc., Santa Clara, CA, USA), revealed a 6.1 Mb gain in the 16p13.3 region [Figure 2]. This region comprising 146 OMIM genes include critical gene CREBBP. Chromosome analyses of both parents were normal, considering de novo change in the child.

- Facial features (hypertelorism, sparse eyebrows and eyelashes, broad nasal bridge and tip, bilateral low set cup shape ears, bilateral ptosis, long and prominent philtrum, downturned corner of mouth, micrognathia).
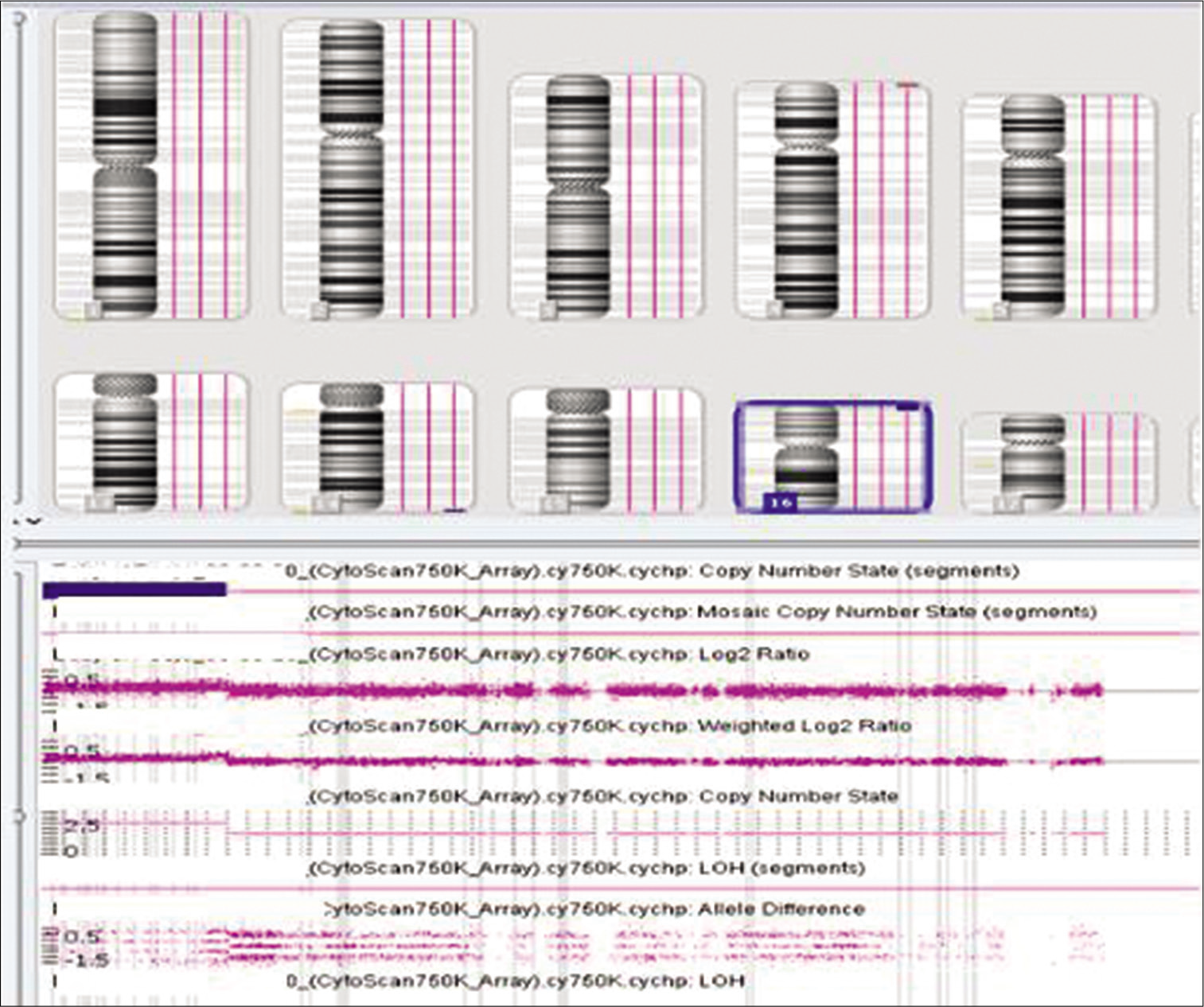
- Array comparative genomic hybridization finding of gain over 16p.
DISCUSSION
The cytogenomic array CGH analysis showed a gain involving chromosome 16p13.3, indicating duplication in this region. The region overlaps the previously described 16p13.3 duplication syndrome and contains the 146 OMIM genes including critical gene CREBBP. Features described in previously affected individuals include growth and mental retardation, specific facial appearance with round head, upslanting and narrow palpebral fissures, sparse eyebrows, broad nasal bridge, rounded nasal tip, prominent glabella, long philtrum, thin upper lip, and micrognathia that changes with age, palatal defects, digital, and musculoskeletal anomalies (club feet, congenital hip dislocation, or camptodactyly of the fingers and toes), anomalies of the heart (ASD, tetralogy of Fallot), and urogenital system.[5,6] The facial features of our patient were similar to those of the reported cases. The previous studies demonstrated that the phenotypic severity could not be related to the size of the duplicated segment.
In our patient, ASD of 3 mm was present. Patients with large 16p duplications have been reported with ASD, tetralogy of Fallot or ventricular septal defect or early onset of pulmonary vascular disease.[7,8] Thus, a baseline cardiological evaluation and periodic monitoring for the potential onset of pulmonary hypertension are recommended.
Seizures and spasticity were present in our case without structural CNS anomalies. McDigillo et al. described two cases with duplication 16p along with review of previously published nine cases. They found neurological manifestations such as seizures in 5/11 cases, corpus callosum hypoplasia in 3/8 cases, and microcephaly in 6/11 cases.[2] The urinary anomalies described in the literature were not found in our case.[9]
This variability of symptoms may present need of characterization of the phenotypic variation of such genetic syndromes at different deletion sizes to improve the clinical diagnosis and management. Microarray offers a genome-wide analysis allowing detection of deletions and duplications thus improving the yield of diagnostic test.
Early diagnosis of 16p duplication in pediatric patients with multiple congenital anomalies helps to optimize a multidisciplinary treatment approach. Delay in diagnosis could lead to financial burden and unfavorable outcomes. The confirmation of diagnosis is crucial for prognostication, and to provide recurrence risk for future pregnancy in the family.
CONCLUSION
We have confirmed the existence of a rare genetic syndrome in a child with duplication 16p, involving the 13.3 region. Facial dysmorphism, growth and mental deficiency, digital, and heart abnormalities are clinical clues for suspecting this rare syndrome, which can be confirmed by newer molecular cytogenetic testing methods. Neurologic deficit such as seizures and spasticity, as occurring in our case, can be a feature of dup16p. Increased awareness of the advanced genomic technologies such as array CGH may enable clinicians to diagnose such disorders and provide prognosis and course of disease and for preconceptional genetic counseling.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Duplication of the Rubinstein-Taybi region on 16p13.3 is associated with a distinctive phenotype. Am J Med Genet A. 2008;146A:2313-7.
- [CrossRef] [PubMed] [Google Scholar]
- 16p subtelomeric duplication: A clinically recognizable syndrome. Eur J Hum Genet. 2009;17:1135-40.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular rulers' for calibrating phenotypic effects of telomere imbalance. J Med Genet. 2002;39:734-40.
- [CrossRef] [PubMed] [Google Scholar]
- Trisomy of chromosome 16p13.3 due to an unbalanced insertional translocation into chromosome 22p13. Eur J Med Genet. 2005;48:355-9.
- [CrossRef] [PubMed] [Google Scholar]
- Duplication 16p13.3 and the CREBBP gene: Confirmation of the phenotype. Eur J Med Genet. 2013;56:26-31.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term follow-up of a 26-year-old male with duplication of 16p: Clinical report and review. Am J Med Genet A. 2007;143A:399-408.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary hypertension and trisomy 16. Pediatr Cardiol. 1998;19:187-9.
- [CrossRef] [PubMed] [Google Scholar]
- Five familial cases with a trisomy 16p syndrome due to translocation. Clin Genet. 1979;16:205-14.
- [CrossRef] [PubMed] [Google Scholar]
- 16p subtelomeric duplication with vascular anomalies: An Albanian case report and literature review. Balkan J Med Genet. 2012;15:73-6.
- [CrossRef] [PubMed] [Google Scholar]




