
Translate this page into:
Glycated hemoglobin and subsequent risk of microvascular and macrovascular complications
*Corresponding author: Saptadip Samanta, Department of Physiology, Midnapore College, Raja Bazar, Midnapore - 721 101, West Bengal, India. saptadip174@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Samanta S. Glycated hemoglobin and subsequent risk of microvascular and macrovascular complications. Indian J Med Sci 2021;73(2):230-8.
Abstract
Diabetes mellitus (DM) is a global health problem and its prevalence is constantly increasing over the past several decades. Measurement of glycated hemoglobin (HbA1c) is widely accepted as indicator of long-term glycemic exposure and used as tool for the diagnosis and management of DM. Patients with DM are at heightened risk of both microvascular and macrovascular complications which affect the several organs including skin, heart, brain, eyes, and kidneys. There is a common etiology between hyperglycemia and vascular diseases. The mechanism of pathogenesis starts with formation and accumulation of advanced glycation end product, impaired vasodilatory response, smooth muscle cell dysfunction, overproduction of endothelial growth factors, chronic inflammation, hemodynamic dysregulation, impaired fibrinolytic activity, and enhanced platelet aggregation. These events decrease the diameter of the vessel and initiate the lesion of the vessel wall followed by local ischemia and tissue damage.
Keywords
Diabetes mellitus
Glycated hemoglobin
Microvascular
Macrovascular diseases

INTRODUCTION
In the 21st century, the prevalence of diabetes mellitus (DM), particularly type 2 diabetes, is a great challenge in our modern society. A report indicated that the number of affecting people is growing day by day and it will be reached about 439 million in 2030, 7.7% of the world’s adult population.[1] In general, fasting blood glucose test is the normal practice for diabetes management, but it indicates blood sugar level only at the moment of the test. The fasting blood glucose level does not give any indication of the previous sugar status. Moreover, it is sometimes influenced by food intake over the previous 12 h. This gap is filled by measuring the glycated/ glycosylated hemoglobin (HbA1c).
The red blood cells (RBCs) of all individuals contain hemoglobin (Hb), which is responsible for carrying oxygen to the tissues through the blood circulation. When Hb combines with glucose (sugar), the molecule called glycosylated (or glycated) Hb (Hb A1c or HbA1c). This form of Hb shows the picture of average plasma glucose concentration over a prolonged period of time. HbA1c forms through non-enzymatic process in all individuals and its normal range usually varies between 3 and 5% of blood.[2] Hb A1c was first separated from other forms of Hb by Allen et al.[3] The relationship between DM and HbA1c was first described in 1969 by Rahbar et al.[4] The use of Hb A1c for monitoring the degree of control of blood glucose in hyperglycemic patients was proposed by Koenig et al.[5] The amount of HbA1c in RBCs is proportional to the concentration of glucose in the blood and to the age of the RBCs. The average life span of RBC is about 120 days. In hyperglycemic patient, the level of HbA1c remains high and persists up to the age of RBC. Thus, HbA1c gives the average percentage of blood glucose level over the previous 2–3 months. The diabetogenic HbA1c has been strongly associated with microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular (ischemic heart disease, peripheral vascular disease, and cerebrovascular disease) complications and starts organ and tissue damage in approximately one-third to one half of the affected people.[6] Measurement of HbA1c is treated as predictor of cardiovascular events.[7] The current review has been focused on formation of HbA1c and pathogenesis of microvascular and macrovascular complications.
FORMATION OF HbA1c
The glycosylation of Hb occurs within the erythrocytes as the glucose entry is facilitated by the glucose transporter1 (GLUT1). Basically, GLUT1 is not insulin dependent, shows very high affinity for glucose, and also maintains constant glucose supply in hypoglycemic condition. RBC is fully housed with Hb (chromoprotein), composed by heme (non-protein) and globin (protein). Heme is an iron-containing protoporphyrin IX and globin contains two alpha and two beta chains. The N-terminal end of beta-chain of adult Hb carries valine. The free α-amino group of valine is glycated in presence of glucose. The pKa values of α-amino group of N-terminal valine1 are lower than the ε-amino group of lysine (close to valine) of beta-chain of Hb. Thus, valine shows more affinity to glucose in normal blood pH (7.4). Moreover, positively charged cavity around the Val1 attracts the sugar molecules for binding.[8] Thus, the post-synthetic glycosylation is a non-enzymatic process which depends on the concentration of plasma glucose levels.
The mechanism of glycation is called Maillard reaction, in which a labile Schiff-base compound is initially formed followed by Amadori rearrangement and finally gives Amadori product; here, it is HbA1c [Figure 1].[2,9] Although it has been predicted that HbA1c correlates with average blood sugar level over the preceding 3 months,[10] there is some controversy. Actually, the circulatory erythrocytes are not in same age at a time. The glycosylation process has been done in older erythrocytes; however, the numbers of younger erythrocytes are more and the average half-life is 30 days.[11] Thus, it has been assumed that HbA1c value contributes 50% of result from 30 days before sampling and only 10% value reflects the previous 90–120 days.[12] There are multiple factors those affect HbA1c levels in addition to blood glucose concentration. These include erythrocyte membrane permeability, genetics (high glycators or low glycators), regular aspirin intake, oxidative stress, rate of erythropoiesis, renal failure (uremic patient), rate of hemolysis, chronic liver disease, and drugs (e.g., ribavirin and dapsone).[2] Hemolysis, recent blood transfusion, acute blood loss, hypertriglyceridemia, and certain drugs inappropriately decrease HbA1c level; while deficiency of iron and Vitamin B12, hyperbilirubinemia, uremia, alcoholism, and few drugs increase HbA1c levels.[13] Drugs such as dapsone, ribavirin, antiretrovirals, and trimethoprim-sulfamethoxazole increase the rate of hemolysis and falsely lower the HbA1c level. These drugs reduce the lifespan of erythrocyte, thereby increasing the proportion of circulatory younger RBC, where rate of glycation is low. Genetic variants of Hb (e.g., HbS trait and HbC trait), Hb derivatives (e.g., carbamylated Hb in patients with renal failure or acetylated Hb in patients taking large amounts of aspirin), recovery from acute blood loss, and hemolytic anemia give impair result of glycated Hb. Myeloproliferative disorders treating drug hydroxyurea acts as antimetabolite which promotes the shifting of the Hb pattern from HbA to HbF. The result is apparent decrease in HbA1c levels. High dose of Vitamins C and E reduces the rate of glycation of Hb due to their strong antioxidant property and falsely lower the test result. Similarly, treatment with erythropoietin falsely lowers the HbA1c values. Deficiency of iron and Vitamin B12 is associated with anemia and reduces the number of younger RBC in circulation which causes high HbA1c levels. Chronic opiate use can increase HbA1c levels.[13] Thus, the measurement of HbA1c does not always reliable. Alternatively, fructosamine assay and quantification of glycated albumin have been considered to provide information about blood sugar level for a short period.[12]
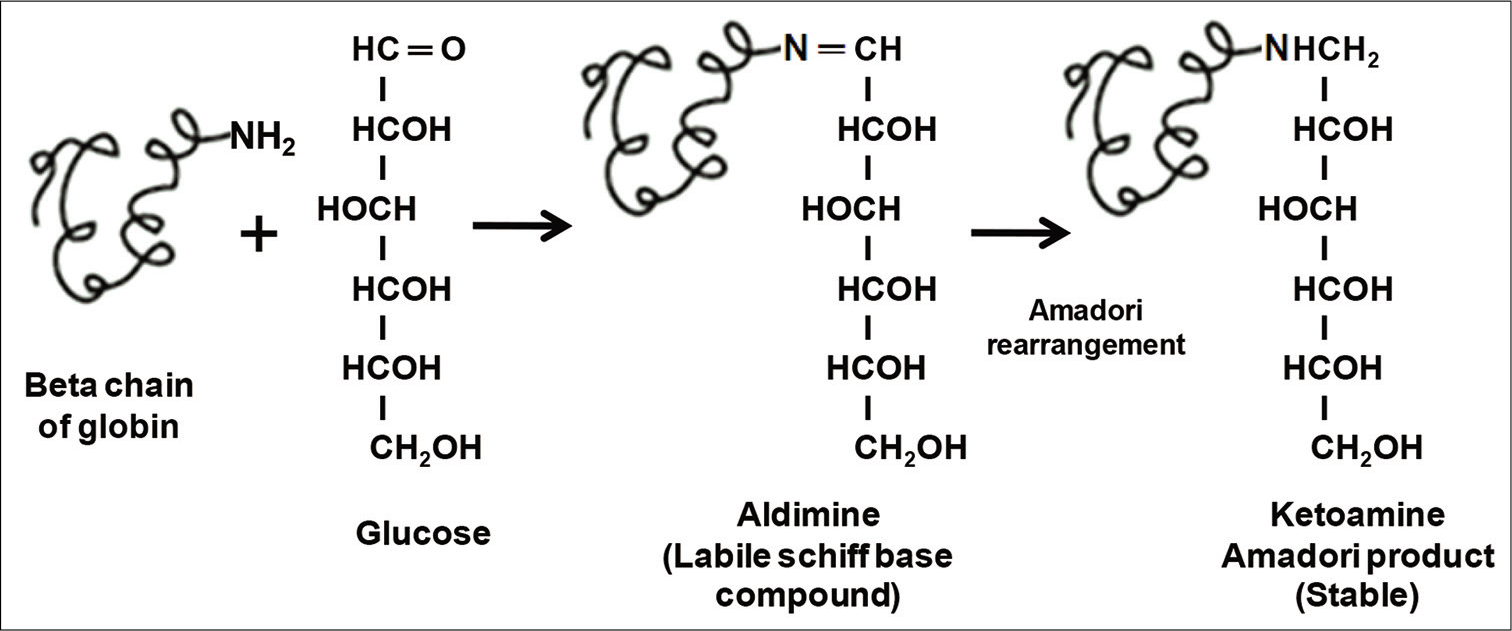
- Mechanism of non-enzymatic glycosylation of hemoglobin.
REFERENCE VALUES AND MEASUREMENT OF HbA1c
The value of HbA1c reflects the average level of plasma glucose concentration over the previous 2–3 months during the life cycle of RBCs (100–120 days).[8] The greater value has been observed in DM. The American Diabetes Association (ADA) and World Health Organization have recommended that the cutoff point HbA1c will be restricted in ≥6.5% for diagnosis of diabetes.[14,15] The ADA also recommended that patients with HbA1c value in between 5.7% and 6.4% are pre-diabetic.[14]
HbA1c test is routinely utilized for long-term assessment of glycemic control. At present, several methods have been applied for the measurement of HbA1c in clinical laboratories. These methods are cation exchange-high-performance liquid chromatography (CE-HPLC), boronate affinity high-performance liquid chromatography (BAC), capillary electrophoresis, and immunoassay.[16,17] However, the level of HbA1c is falsely affected by rate of hemolysis, life span of RBC, presence of Hb variants (HbC, HbS, HbE, HbD, and HbF), and elevated level of modified Hb (carbamylated, acetylated, and labile HbA1c).[13,18]
HbA1c carries electrochemical charges on their surface which differs from the charges of other components of Hb. The molecular size of HbA1c is different from other components. These two factors (charge and size) are used to detect HbA1c.[16] Three main techniques are applied for the measurement of HbA1c in clinical system. These include (i) chromatography-based HPLC assay, (ii) antibody-based immunoassay, and (iii) enzymatic assay. In general, HPLC technique was the first automated systems to measure the total HbA1c level. The measurement process is based on the ratio of HbA1c peak area to the total Hb peak areas.[19] Affinity chromatography can also be applicable for the detection of HbA1c. In this purpose, boronate is used as adsorbent because it has unique features, specifically binds with cisdiol configuration of stable glucose of HbA1c. The method can detect the all stable forms of HbA1c species.[16] The latex coated immunoassay method is based on agglutination technique. The HbA1c-specific antibodies are coated on latex beads which bind with HbA1c (antigen). The antigen-antibody reaction alters the turbidity of the reaction mixture which is proportional to the amount of the antigen (HbA1c) in the samples.[20] Recently, direct enzymatic assay method has been developed to measure HbA1c. A recombinant fructosyl valine oxidase (FVO) is used for reaction purpose which recognizes glycated valine as substrate. The initial step of cell lysis is done by lysis buffer which contains oxidizing agents to discard low-molecular-weight and high-molecular-weight signal interfering molecules. After lysis, the whole blood samples are subjected to proteolytic digestion for release of amino acids including glycated valines. The recombinant FVO reacts with N-terminal glycated valines and then produces hydrogen peroxide. The production of hydrogen peroxide is directly proportional to the concentration of glycated valines. The level of hydrogen peroxide is measured in the presence of horseradish peroxidase and a suitable chromagen.[21]
INDICATION OF HEALTH STATUS AND HbA1c
The ADA has recommended routine measurement of HbA1c at least twice annually for diabetic patients whose blood sugar is well-controlled and more frequently in those has persistently elevated blood sugars. In this purpose, ADA has proposed “ABCs of Diabetes Program” which indicates monitoring of A1c and measurement of blood pressure and cholesterol. Persistent elevations in blood sugar increase the risk long-term vascular complications such as coronary disease, heart attack, stroke, heart failure, kidney failure, blindness, erectile dysfunction, neuropathy (especially in the feet), and gangrene. Thus, measurement of HbA1c can predict the risk of developing many of these chronic complications associated with diabetes. Mutational disorders affect the HbA1c levels. Low values of HbA1c than the expected result have been seen in the person with glucose-6-phosphate dehydrogenase deficiency or sickle cell anemia as the life span of RBC is short in these conditions. However, nutritional deficiency of Vitamin B12 and folic acid increases the level of HbA1c than the expected values.
RELATIONSHIP BETWEEN HbA1c AND MICROVASCULAR AND MACROVASCULAR COMPLICATIONS
Microvascular and macrovascular complications occur concomitantly and the risk is high in type 2 diabetic patients. Several authors reported that microvascular diseases are associated with type 2 diabetes which increases the risk of cardiovascular disease (CVD) and death.[22-24] Sorensen et al.[25] reported that type 2 diabetic patients are more prone to retinal and skin microvascular abnormalities compared to pre-diabetic and susceptible to the development of CVD. The recent report indicated that microvascular disease alone plays an important role in the development of diabetic angiopathy.[22] The common risk factors of microvascular diseases are age, prevalence of diabetes, tobacco use, dyslipidemia, and hypertension.
Pathophysiology of microvascular and macrovascular diseases
DM alters the structural and functional characteristics of vascular bed. The microvascular diseases are associated with damage of tiny vessels including capillaries. Macrovascular complications are related to injury in large vessels, such as arteries and veins. The pathogenesis of both microvascular and macrovascular diseases has same etiological characteristics. The relationship between microvascular and macrovascular diseases is multifactorial (metabolic and structural disintegrity) and associated with DM. The mechanism of pathogenesis is schematically given in Figure 2. Prolonged hyperglycemic condition is the initiator of microvascular complications. The cell cannot use excess glucose; the result is activation of polyol pathway, formation of sorbitol. Actually, the metabolism of glucose starts after activation of hexokinase enzyme; but, the enzyme is saturated during hyperglycemic condition. Thus, excess glucose activates nicotinamide adenine dinucleotide phosphate (NADPH)-dependent aldose reductase which converts the glucose to sorbitol. In general, sorbitol is not freely permeable through the cell membrane. Sorbitol binds with matrix protein, forms hexosamine compounds called advanced glycation end products (AGEs), which increases the expression of its own receptor. The ligand receptor complex starts signal transduction for activation of protein kinase C (PKC)-mediated pathway and ultimately promotes overproduction of reactive oxygen species (ROS) in mitochondria. On the other hand, synthesis of sorbitol decreases the amount of NADPH which is the essential component for regeneration of activate glutathione, a potent cellular antioxidant. The synergistic effect is the generation of oxidative stress which is associated with vascular diseases.[26,27] Oxidative stress lowers nitric oxide (NO) synthesis, inhibits vasodilation, and also induces cell proliferation, vascular hypertrophy, apoptosis, and inflammation in endothelial cells and smooth muscle of the vascular wall.[28-30]
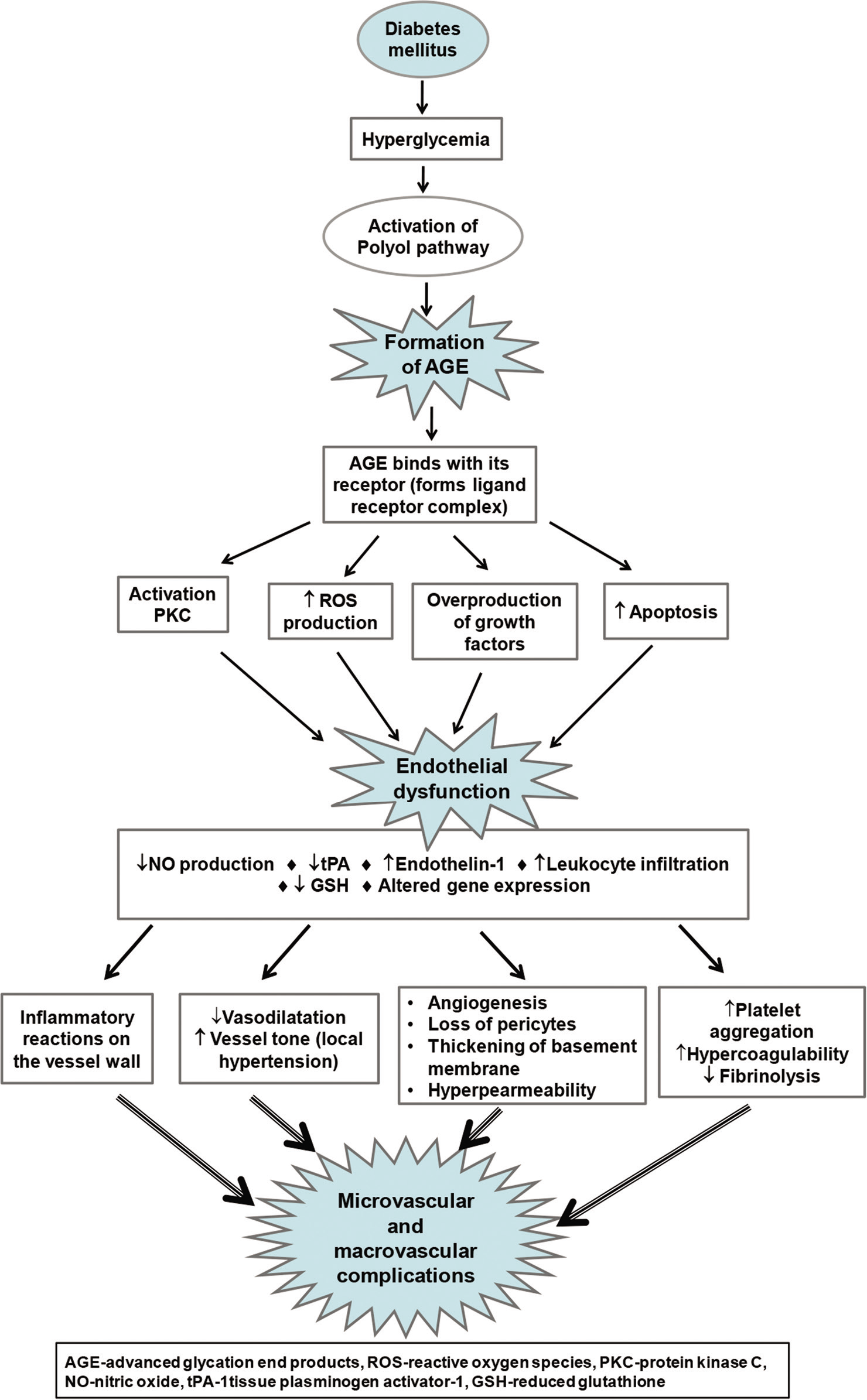
- Potential mechanisms of pathogenesis for diabetes-associated endothelial dysfunction and progression of microvascular and macrovascular disease.
Hirakawa et al.[31] reported that visit-to-visit glycemic variability in HbA1c as well as that in fasting glucose is the predictor to assess the future development of macrovascular and microvascular events. Although there is a relationship between hyperglycemia and HbA1c values, the value of fasting glucose is more significantly associated with vascular complications. Actually, chronic hyperglycemia or long-term glucose variability is measured by HbA1c, while continuous glucose monitoring gives the picture of short-term (day to day) glycemic variability.[32] Thus, daily glucose variability and fasting plasma glucose levels are the stronger predictors than those of HbA1c. Ohara et al.[32] had demonstrated the relationship between oxidative stress and day-to-day glucose variability in type 2 diabetic patients. Visit-to-visit glucose fluctuation increases oxidative stress by generating overproduction of superoxide.[31] High level of oxidative stress increases inflammatory cytokines levels and induces cellular apoptosis and endothelial damage.
In addition, high blood glucose levels accelerate macrophage adhesion to endothelial cells which promote the formation of fibrotic arteriosclerotic lesions. The subsequent result is progression of atherosclerosis.[31-35]
Besides these, other complications such as insulin resistance, metabolic syndrome, hypertension, dyslipidemia, and obesity are also influenced by oxidative stress.[27,36] The depletion of endothelial progenitor cells (EPCs) is also associated with vascular diseases. EPCs arise in bone marrow and circulated through blood. These cells appear as immature cells and have capacity to differentiate into mature endothelial cells. They are characterized by simultaneous expression of CD34+, CD133+, and vascular endothelial growth factor receptor 2 (VEGF 2). EPCs are essential for endogenous endothelial repair and wound healing. The number of EPCs diminishes during vascular dysfunction. Reduced number and impaired functions of EPCs have been observed in microvascular and macrovascular disease, particularly in CVD.[37,38] Moreover, microvascular disease in diabetic patients increases the chances atherosclerosis.[39] Thus, consequent events alter the characteristics, tone, flexibility, thickness, and intracellular biochemical activities of capillary or vascular wall, leading to the progression of microvascular and macrovascular diseases.
Microvascular complications
Diabetic retinopathy (DR)
DR is a common microvascular complication, leads to visual disability and blindness. This disease affects the peripheral part of the retina, the macula, or both. In the proliferative stage of DR, abnormal growth of new vessels maximally damages the eye. The hemorrhage in vitreous humor or retinal detachment is the major cause of total or partial vision loss. Moreover, central vision loss can occur through retinal vessel leakage and subsequent macular edema.[40] Hypertension and obesity also accelerate the chances of DR in diabetic patients.[41] The endothelial cells of small vessels are surrounded by pericytes, elongated contractile cells.[42] These contractile cells maintain the capillary tone and protect the capillary against ROS-mediated damage.[43] Oxidative stress wipe outs the pericytes. Dilation and formation of new capillary do not possible when pericytes are lost. Capillary basement membrane is one of the important parts of capillary as it holds the endothelial cells and loose-connective tissue in proper position. In addition, basement membrane is essential for angiogenesis. Thickening of capillary basement membrane weakens the vessel walls and increases permeability of endothelial cells.[6] Hyperglycemia impairs retinal blood flow which stimulates the inflammatory response over the retinal blood vessels. The result is occlusion of capillary and initiation of hypoxia-induced retinal damage.[44]
The mechanism of pathogenesis in DR is associated with the formation of AGEs. Hyperglycemia promotes the formation of sorbitol followed by AGEs. AGE increases collagen content and thickness of basement membrane at the capillary wall.[45]
Actually, AGE activates PKC, stimulates ROS production, and promotes several cellular events such as apoptosis of retinal pericytes,[46] overproduction of VEGF, insulin-like growth factor 1 (IGF1), fibroblast growth factor (FGF) and hepatocyte growth factor,[47] angiogenesis, and exaggeration of inflammatory response on the vascular wall.[48] These impaired functions increase the risk of plaque formation, capillary blockage, and retinal ischemia.[49] Angiogenesis is the step of neovascularization. Loss of pericytes, damage of basement membrane, and hyperpermeability of endothelial cell layer elevate the risk of hemorrhage towards vitreous humor. VEGF and FGF start retinal fibrosis and detachment, leading to vision loss.[40]
Diabetic neuropathy
Peripheral neuropathy (PN) is another disorder for the people with DM. High stature, presence of CVD, metabolic ketoacidosis, and microalbuminuria increase the risk of PN.[50] Pain and loss of sensation followed by ulceration in lower extremity are the common features of PN.[51] Sometimes, autonomic neuropathy may create cardiovascular dysfunctions such as abnormal heart rate and blood pressure.[52] The effects of PN start due to peripheral vascular dysfunction. The initial stage of pathogenesis starts with reduced blood flow in the capillary bed near the nerve endings which initiates thickening of basement membrane, loss of pericyte, and damage of microfilaments. All these effects lower the rate of perfusion at the nerve terminal and create endoneurial hypoxia.[6] Hyperglycemia influences the production of AGE as similar as DR. The high glycemic index induces neuronal microvascular damage which activates signal cascade of demyelination as well as PN.[53] Long-term exposure to high glycemic variability starts glycosylation on the PO protein in myelin of peripheral nerves. PO is a glycoprotein, present in myelin sheath, termed as myelin protein zero. Exposure to glucose with PO forms AGE adducts on peripheral nerve myelin. The irreversible myelin AGE adduct is recognized by macrophages. This interaction starts inflammatory response followed by initiation of segmental demyelination. After degeneration of nerve fibers, the rate of regeneration is very slow because accumulated AGE activates PKC pathways and induces oxidative stress-mediated damage.[54] Diabetogenic demyelination and impaired regeneration finally accelerate PN.
Diabetic nephropathy
DM progressively increases the rate of renal dysfunction. Microalbuminuria is the indicator of initiation renal dysfunction and albuminuria occurs at severe stage, finally renal failure.[55] During disease progression, glomerular basement membrane thickness and filtration rate increase. The result is albumin excretion in urine.[56] Formation of AGE during hyperglycemic condition has crucial role in pathogenesis. AGE increases apoptotic rate of mesangial cells, thickness of basement membrane, alters the characteristics of podocytes, and alters the characteristics of extracellular matrix proteins, leading to glomerular hyperfiltration and sclerosis.[57] Oversecretion of IGF1 and transforming growth factor β is mediated by AGE; both these agents contribute key role in glomerular fibrosis. In addition, excess glucose is converted to sorbitol through polyol pathway which increases osmotic pressure at the glomerular filtrating bed and damages the endothelium. Synthesis of sorbitol also changes the redox homeostasis at the surrounding area, leading to oxidative stress-induced further tissue damage.
Macrovascular complications
Cardiovascular disease
About 70% of type 2 diabetic patient are suffering for CVD. Diabetic people have 5-fold greater risk of myocardial infarction which ultimately increases the risk of congestive heart failure and death.[58] Commonly diabetic patients have impaired lipid profile (elevated serum triglyceride, low-density lipoprotein [LDL], and free fatty acid levels and low levels of high-density lipoprotein). These factors are associated with CVD. Beckman et al.[59] reported that hypertension, hyperglycemia, dyslipidemia, and chronic inflammation damage the vascular endothelial cell lining, leading to macrovasculopathy and CVD.
DM initially alters the activity of vascular endothelium which gradually progresses to macrovascular diseases. Uncontrolled blood sugar level as well as AGE inhibit the activity of NO.[60] NO is a potent vasodilator and maintains the arteriolar diameter. de Vriese et al.[61] reported that hyperglycemia inhibits endothelial NO synthase (eNOS) and increases the production of ROS, leading to further inhibition of NO synthesis. NO has several functions like activation of tissue plasminogen activator which converts plasminogen to plasmin, a potent fibrinolytic enzyme. This is essential for breakdown of microthrombotic clot within the small vessel. NO-mediated vasodilatation also prevents adherence of inflammatory cells on the endothelial surface.[62] Insulin resistance type 2 diabetes increases the release of free fatty acids from adipose tissue. High blood glucose level and free fatty acids stimulate ROS production through PKC-dependent activation of NADPH oxidase in the vascular smooth muscle cells and endothelial cells.[63] Therefore, inhibition of eNOS and ROS-mediated oxidative stress promotes vascular damage and accelerates atherosclerosis. Furthermore, vasoconstrictor substances play a crucial role in macrovascular complications. Endothelin 1 is one of the local vasoconstrictor agents, which exerts vasoconstrictive effects. The result is retention of salt and water within the vessel. This effect increases the pressure on the vessel wall. Moreover, retention of salt and water also activates rennin angiotensin system in kidney.[64] Hyperglycemia also impairs platelet functions, which increases the risks for thrombus plaque formation in vessel.[62] All these effects increase the chance of intravascular coagulation and lower the fibrinolytic capacity. The consequence is vascular damage and initiation of inflammatory response. Inflammatory cells enter within the damage area and migrate into the deeper layers of vessel. These cells are converted to foam cells after ingestion of oxidized LDL.[65] Foam cells form the central core of the atherosclerotic fatty streaks; the result is occlusion of vessels.[6,66] Hyperglycemia is not only the key factor for progressive vascular damage; other two factors such as hypertension and dyslipidemia are also strongly associated with cardiovascular events in patients with type 2 diabetes.[67] The diabetic patient shows medial vascular calcification or Mönckeberg medial sclerosis which is the most important risk factors for CVD.[68] Thus, the central pathological mechanism of CVD is the narrowing of the vessels and appearance of local ischemic condition. This happens in response to endothelial injury, inflammation, accumulation of oxidized lipids from LDL, formation of foam cells, infiltration of monocytes and macrophages, activation of T-lymphocytes, and synthesis of collagen.[69] The result is lesion within the vessel wall which starts acute vascular infarction followed by coronary ischemic disease.
Cerebrovascular disease
Diabetes increases cerebrovascular stroke up to 2–4-fold.[6] Diabetogenic hyperglycemia affects the cerebrovascular circulation which has greater risk of intracranial and extracranial atherosclerosis.[70] Diabetes raises the chronic inflammatory substances in blood which increases the risk of vascular damage and stroke.[71] The mechanism of pathogenesis of cerebrovascular diseases is similar to CVD. Hyperglycemia, high HbA1c levels, low expression of NO synthase, and high levels of AGE are associated with endothelial dysfunction, impaired vasodilatation, free radical formation, and impaired cerebral autoregulation. These factors alter the cerebrovascular activity. As a result, blood vessel walls become fragile and weaken at the blood–brain barrier, which increases the risk of brain edema and reperfusion injury.[72] The cerebrovascular dysfunction also produces local ischemia and intracellular acidosis which affects the neurons and glial cells.[73] Intracellular acidosis disrupts the normal cellular homeostasis and induces ROS generation and DNA damage. The glycogenic amino acid glutamate remains high during hyperglycemic condition. Glutamate acts as excitatory neurotransmitter which promotes influx of calcium within the neurons. High levels of intracellular calcium initiate metabolic disorders by activating different enzymes such as lipase, protease, endonuclease, kinases, and phosphatases. Calcium overload also enhances PKC-mediated ROS production. These effects have role on mitochondrial injury and cell death. ROS-mediated free radicals are the key initiator of microvascular dysfunction that advances the upregulation of intercellular adhesion molecule 1, microvascular plugging, and inflammatory response at the endothelial cell surface. Thus, calcium is a mediator of cerebrovascular damage which may lead to ischemic brain disease in people with diabetes.[74]
Peripheral arterial disease (PAD)
PAD is characterized by occlusion of vessels mostly at the lower extremity. Ankle-brachial index is the measure of PAD.[75] Diabetes, along with elevated serum fibrinogen levels, dyslipidemia, and physical inactivity, is the major causes of PAD.[76] The symptoms of the disease are cramping, pain followed by ulceration, and disability in the lower extremity.[6]
CONCLUSION
Glycosylated Hb gives the picture of blood sugar concentration of the previous 2–3 months and also helps to insight data about blood sugar control in patients within treatment procedure. High blood sugar increases the level of HbA1c as well as activation of polyol pathway and formation of AGEs. AGE is the main factor for pathogenesis of microvascular and macrovascular diseases. AGE alters the characteristics of vascular wall, increases oxidative stress, and inflammatory response; the consequences are damage of vessel wall, local ischemia, and progression of complications.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4-14.
- [CrossRef] [PubMed] [Google Scholar]
- Current controversies in the use of haemoglobin A1C. J Intern Med. 2012;271:227-36.
- [CrossRef] [PubMed] [Google Scholar]
- Observations on the chromatographic heterogeneity of normal adult and fetal human hemoglobin: A study of the effects of crystallization and chromatography on the heterogeneity and isoleucine content. J Am Chem Soc. 1958;80:1628-34.
- [CrossRef] [Google Scholar]
- Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem Biophys Res Commun. 1969;36:838-43.
- [CrossRef] [Google Scholar]
- Correlation of glucose regulation and hemoglobin AIc in diabetesmellitus. N Engl J Med. 1976;295:417-20.
- [CrossRef] [PubMed] [Google Scholar]
- Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys Ther. 2008;88:1322-35.
- [CrossRef] [PubMed] [Google Scholar]
- Haemoglobin A(1c) levels and subsequent cardiovascular disease in persons without diabetes: A meta-analysis of prospective cohorts. Diabetologia. 2011;54:1327-34.
- [CrossRef] [PubMed] [Google Scholar]
- Review of hemoglobin A(1c) in the management of diabetes. J Diabetes. 2009;1:9-17.
- [CrossRef] [PubMed] [Google Scholar]
- Translating the A1C assay into estimated average glucose values. Diabetes Care. 2008;31:1473-8.
- [CrossRef] [PubMed] [Google Scholar]
- Diabetes control and complications: The role of glycated haemoglobin, 25 years on. Diabetes Med. 2004;21:657-65.
- [CrossRef] [PubMed] [Google Scholar]
- Drugs affecting HbA1c levels. Indian J Endocrinol Metab. 2012;16:528-31.
- [CrossRef] [PubMed] [Google Scholar]
- Classification and diagnosis of diabetes. Diabetes Care. 2017;40:S11-24.
- [CrossRef] [PubMed] [Google Scholar]
- Use of Glycated Haemoglobin (HbA1c) in the Diagnosis of Diabetes Mellitus Geneva, Switzerland: World Health Organization; 2011. p. :1-25.
- [Google Scholar]
- False measurement of glycated hemoglobin in patients without hemoglobin A. Biosci Rep. 2019;39:BSR20180128.
- [CrossRef] [PubMed] [Google Scholar]
- Hereditary spherocytosis and other hemolytic anomalies distort diabetic control by glycated hemoglobin. Clin Lab. 2006;52:477-81.
- [Google Scholar]
- Conditions associated with very low values of glycohaemoglobin measured by an HPLC method. J Clin Pathol. 2004;57:346-9.
- [CrossRef] [PubMed] [Google Scholar]
- Quantitative measurement of HbA1c by an immunoturbidimetric assay compared to a standard HPLC method. Am J Clin Pathol. 1995;104:89-95.
- [CrossRef] [PubMed] [Google Scholar]
- Direct enzymatic assay for %HbA1c in human whole blood samples. Clin Biochem. 2008;41:576-83.
- [CrossRef] [PubMed] [Google Scholar]
- Comparative effects of microvascular and macrovascular disease on the risk of major outcomes in patients with Type 2 diabetes. Cardiovasc Diabetol. 2017;16:95.
- [CrossRef] [PubMed] [Google Scholar]
- The influence of baseline risk on the relation between HbA1c and risk for new cardiovascular events and mortality in patients with Type 2 diabetes and symptomatic cardiovascular disease. Cardiovasc Diabetol. 2016;15:101.
- [CrossRef] [PubMed] [Google Scholar]
- Microvascular disease and risk of cardiovascular events among individuals with Type 2 diabetes: A population-level cohort study. Lancet Diabetes Endocrinol. 2016;4:588-97.
- [CrossRef] [Google Scholar]
- Prediabetes and Type 2 diabetes are associated with generalized microvascular dysfunction: The Maastricht study. Circulation. 2016;134:1339-52.
- [CrossRef] [PubMed] [Google Scholar]
- Increased cardiovascular morbidity and mortality in Type 2 diabetes is associated with the glutathione S transferase theta-null genotype: A Go-DARTS study. Circulation. 2005;111:2927-34.
- [CrossRef] [PubMed] [Google Scholar]
- Oxidative stress and diabetic complications. Circ Res. 2010;107:1058-70.
- [CrossRef] [PubMed] [Google Scholar]
- Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73:411-8.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc Diabetol. 2013;12:142.
- [CrossRef] [PubMed] [Google Scholar]
- Oxidative stress, inflammation, endothelial dysfunction and incidence of Type 2 diabetes. Cardiovasc Diabetol. 2016;15:51.
- [CrossRef] [PubMed] [Google Scholar]
- Impact of visit-to-visit glycemic variability on the risks of macrovascular and microvascular events and all-cause mortality in Type 2 diabetes: The ADVANCE trial. Diabetes Care. 2014;37:2359-65.
- [CrossRef] [PubMed] [Google Scholar]
- Relationship between daily and day-to-day glycemic variability and increased oxidative stress in Type 2 diabetes. Diabetes Res Clin Pract. 2016;122:62-70.
- [CrossRef] [PubMed] [Google Scholar]
- Repetitive fluctuations in blood glucose enhance monocyte adhesion to the endothelium of rat thoracic aorta. Arterioscler Thromb Vasc Biol. 2006;26:2275-80.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with Type 2 diabetes. J Am Med Assoc. 2006;295:1681-7.
- [CrossRef] [PubMed] [Google Scholar]
- Relationships between glucose excursion and the activation of oxidative stress in patients with newly diagnosed Type 2 diabetes or impaired glucose regulation. Endocrine. 2010;37:201-8.
- [CrossRef] [PubMed] [Google Scholar]
- A novel component of the metabolic syndrome: The oxidative stress. Nutr Metab Cardiovasc Dis. 2010;20:72-7.
- [CrossRef] [PubMed] [Google Scholar]
- Circulating progenitor cell count predicts microvascular outcomes in Type 2 diabetic patients. J Clin Endocrinol Metab. 2015;100:2666-72.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term prediction of cardiovascular outcomes by circulating CD34+ and CD34+ CD133+ stem cells in patients with Type 2 diabetes. Diabetes Care. 2017;40:125-31.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical update: Cardiovascular disease in diabetes mellitus: Atherosclerotic cardiovascular disease and heart failure in Type 2 diabetes mellitus mechanisms, management, and clinical considerations. Circulation. 2016;133:2459-502.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular understanding of hyperglycemia's adverse effects for diabetic complications. J Am Med Assoc. 2002;288:2579-88.
- [CrossRef] [PubMed] [Google Scholar]
- Retinopathy in subjects with impaired fasting glucose: The NANSY-Eye baseline report. Diabetes Obes Metab. 2007;10:646-51.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular pericytes not only regulate growth, but also preserve prostacyclin producing ability and protect against lipid peroxide-induced injury of cocultured endothelial cells. Biochem Biophys Res Commun. 1993;190:418-25.
- [CrossRef] [PubMed] [Google Scholar]
- Role of blood flow and impaired autoregulation in the pathogenesis of diabetic retinopathy. Diabetes. 1995;44:603-7.
- [CrossRef] [PubMed] [Google Scholar]
- Advanced glycation end products and diabetic complications. Expert Opin Investig Drugs. 2002;11:1205-23.
- [CrossRef] [PubMed] [Google Scholar]
- Receptor-mediated toxicity to pericytes of advanced glycosylation end products: A possible mechanism of pericyte loss in diabetic microangiopathy. Biochem Biophys Res Commun. 1995;213:681-7.
- [CrossRef] [PubMed] [Google Scholar]
- Advanced glycation end productsinduced apoptosis and overexpression of vascular endothelial growth factor in bovine retinal pericytes. Biochem Biophys Res Commun. 2002;290:973-8.
- [CrossRef] [PubMed] [Google Scholar]
- Leukocytes in diabetic retinopathy. Curr Diabetes Rev. 2007;3:3-14.
- [CrossRef] [PubMed] [Google Scholar]
- Increased prevalence of microthromboses in retinal capillaries of diabetic individuals. Diabetes. 2001;50:1432-9.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence of diabetic peripheral neuropathy and its relation to glycaemic control and potential risk factors: The EURODIAB IDDM complications study. Diabetologia. 1996;39:1377-84.
- [CrossRef] [PubMed] [Google Scholar]
- Foot problems in patients with diabetes mellitus In: Pickup J, Williams G, eds. Textbook of Diabetes. London, United Kingdom: Blackwell Science; 1997. p. :1-58.
- [Google Scholar]
- Accumulation of diabetic rat peripheral nerve myelin by macrophages increases with the presence of advanced glycosylation end products. J Exp Med. 1984;160:197-207.
- [CrossRef] [PubMed] [Google Scholar]
- New insights into the mechanisms of diabetic neuropathy. Rev Endocr Metab Disord. 2004;5:227-36.
- [CrossRef] [PubMed] [Google Scholar]
- The early natural history of nephropathy in Type 1 diabetes, II: Early renal structural changes in Type 1 diabetes. Diabetes. 2002;51:1580-7.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenesis of diabetic nephropathy. Rev Endocr Metab Disord. 2004;5:237-48.
- [CrossRef] [PubMed] [Google Scholar]
- Nonenzymatic glycation of mesangial matrix and prolonged exposure of mesangial matrix to elevated glucose reduces collagen synthesis and proteoglycan charge. Kidney Int. 1993;43:853-64.
- [CrossRef] [PubMed] [Google Scholar]
- Impact of diabetes on long-term prognosis in patients with unstable angina and non-Q-wave myocardial infarction: Results of the OASIS (Organization to Assess Strategies for Ischemic Syndromes) Registry. Circulation. 2000;102:1014-9.
- [CrossRef] [PubMed] [Google Scholar]
- Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. J Am Med Assoc. 2002;287:2570-81.
- [CrossRef] [PubMed] [Google Scholar]
- Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through RAGE activation. Clin J Am Soc Nephrol. 2008;3:691-8.
- [CrossRef] [PubMed] [Google Scholar]
- Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963-74.
- [CrossRef] [PubMed] [Google Scholar]
- Diabetes mellitus as a prothrombotic condition. J Intern Med. 2007;262:157-72.
- [CrossRef] [PubMed] [Google Scholar]
- High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939-45.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of disease: Endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46-56.
- [CrossRef] [PubMed] [Google Scholar]
- Inflammation and atherosclerosis. Ann Rev Pathol. 2006;1:297-329.
- [CrossRef] [PubMed] [Google Scholar]
- Inflammatory mechanisms of diabetic complications. Curr Diabetes Rep. 2007;7:242-8.
- [CrossRef] [PubMed] [Google Scholar]
- The relation between hba1c and cardiovascular events in patients with Type 2 diabetes with and without vascular disease. Diabetes Care. 2015;38:1930-6.
- [CrossRef] [PubMed] [Google Scholar]
- Medial vascular calcification revisited: Review and perspectives. Eur Heart J. 2014;35:1515-25.
- [CrossRef] [PubMed] [Google Scholar]
- Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2008;26:77-82.
- [CrossRef] [Google Scholar]
- The prevalence of calcified carotid artery atheromas on the panoramic radiographs of patients with Type 2 diabetes mellitus. Oral Surg Oral Med Oral Pathol Oral Radiol. 2000;89:420-4.
- [CrossRef] [Google Scholar]
- Inflammation-sensitive plasma proteins, diabetes, and mortality and incidence of myocardial infarction and stroke: A population-based study. Diabetes. 2003;52:442-7.
- [CrossRef] [PubMed] [Google Scholar]
- HbA1c (glycated hemoglobin) levels and clinical outcome post-mechanical thrombectomy in patients with large vessel occlusion. Stroke. 2019;50:119-26.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebral metabolic profile, selective neuron loss, and survival of acute and chronic hyperglycemic rats following cardiac arrest and resuscitation. Brain Res. 1999;821:467-79.
- [CrossRef] [Google Scholar]
- Glutamate, calcium, and free radicals as mediators of ischemic brain damage. Ann Thorac Surg. 1995;59:1316-20.
- [CrossRef] [Google Scholar]
- Ethnic differences in peripheral arterial disease in the NHLBI genetic epidemiology network of arteriopathy (GENOA) study. Vasc Med. 2003;8:237-42.
- [CrossRef] [PubMed] [Google Scholar]
- Risk factors for peripheral arterial disease incidence in persons with diabetes: The atherosclerosis risk in communities (ARIC) study. Atherosclerosis. 2005;180:389-97.
- [CrossRef] [PubMed] [Google Scholar]




