
Translate this page into:
Hemophagocytic lymphohistiocytosis following reactivation of Epstein–Barr virus infection – A rare case report
*Corresponding author: C. Sundaram, Department of General Medicine, Thanjavur Medical College and Hospital, Thanjavur, Tamil Nadu, India. sundrinio@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sundaram C, Shriramganesh RT, Sangeetha G. Hemophagocytic lymphohistiocytosis following reactivation of Epstein– Barr virus infection – A rare case report. Indian J Med Sci 2021;73(2):264-5.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a catastrophic condition, leading to the rapid incitement of immune-mediated cells. Asymptomatic primary Epstein–Barr virus (EBV) infection is common in the general population, while EBV causing acute hepatitis and jaundice is uncommon and acute liver failure has been rarely reported. Here, we report a case of fulminant hepatic failure and HLH following the reactivation of EBV infection that was successfully treated with dexamethasone therapy.
Keywords
Hemophagocytic lymphohistiocytosis
Epstein–Barr virus
Acute liver failure
Fulminant hepatitis

INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is characterized by an abnormally regulated immune system leading to multiorgan damage.[1] Primary HLH is either due to ineffective termination of the immune response or failure to remove the antigen that results in continual activation of macrophages and cytotoxic T cells. Pathogenesis of secondary (acquired) HLH is still unclear, although heterozygous changes or polymorphisms in the familial HLH genes are seen in patients with secondary forms of HLH.[2] Herewith, we report an adolescent male who had fulminant hepatic failure and HLH following reactivation of Epstein–Barr virus (EBV) infection who responded to dexamethasone therapy.
CASE REPORT
An 18-year-old previously healthy male presented to us with massive hematemesis and unstable vitals. He had a fever, jaundice, and abdominal distension of 10 days duration. Clinical examination revealed severe anemia, jaundice, hepatosplenomegaly, and asterixis. Laboratory blood results showed Hb – 4.3g/dl, TC-3300*109/L, platelet – 6000*109/L, lymphocytes – 66.6%, B.urea – 52 mg/dl, S.creat – 1.6 mg/dl, RBS – 66 mg/dl, T.Bil-3.7mg/dl, AST – 587U/L, ALT – 120U/L, ALP – 127U/L, LDH – 1612.7U/L, INR – 1.52, retic count – 1.3%, total cholesterol – 225 mg/dl, serum triglyceride – 341 mg/dl, and serum ferritin – 550 ng/ml. Atypical lymphocytes were seen in peripheral blood smear and bone marrow examination. Ultrasound abdomen showed gross hepatosplenomegaly. The patient was diagnosed as HLH based on HLH-2004 diagnostic criteria (five out of eight criteria satisfied). Usual causes of Fulminant Hepatic Failure were ruled out by relevant investigations (Serology for Hepatitis A, B, C, E, Lepto IgM, Dengue IgM was negative). The patient had signs of the previous infection from EBV (IgG EBNA – 63.6U/ml, IgG VCA EBV – 100U/ml, IgM VCA EBV: <10U/ml). EBVPCR was positive in high titers which suggest reactivation of EBV infection. He was conservatively managed with the blood transfusion and liver support. Dexamethasone was started immediately according to the HLH-2004 protocol. The patient’s general condition improved dramatically within 2 weeks and was discharged after a tapering dose of steroids and advised to follow-up.
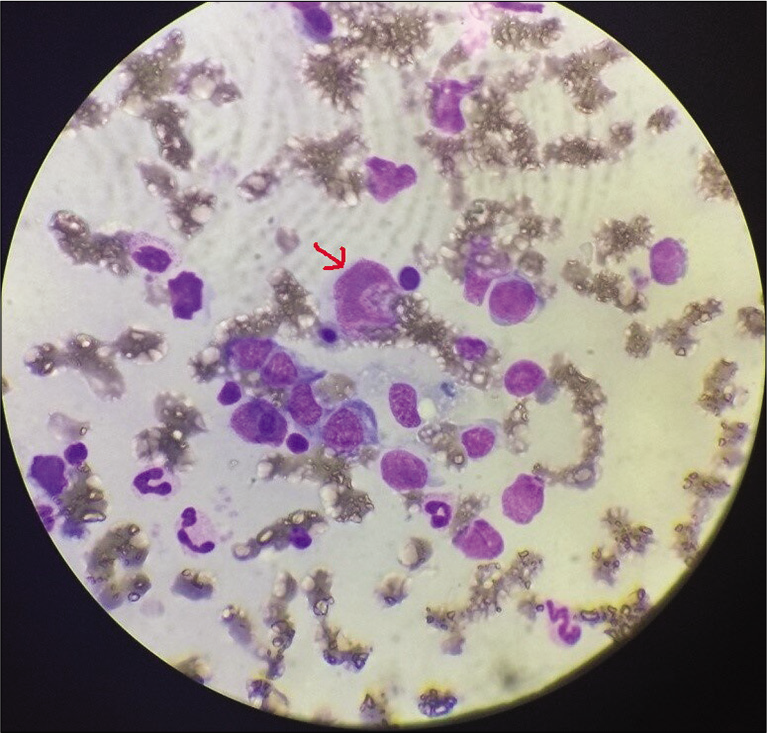
- Bone marrow aspirate showing atypical lymphocytes (arrow).
DISCUSSION
HLH can occur as a primary condition or as a result of the underlying malignancy, infections, and metabolic disorders.[3] EBV is considered the most common cause of infection-associated HLH.[4] Hepatocellular injury is one of the most frequently observed complications. Previous studies involving secondary HLH have shown elevated liver transaminases in almost 85% and hyperbilirubinemia in 50% of patients.[5] Mild-to-moderate elevation of transaminases is seen in most of these cases. Higher transaminase levels reflect the severity of HLH associated with EBV.[4] HLH presenting as liver failure is very rare and often occurs due to multiorgan failure.[6] Hepatocellular steatosis, necrosis, and sinusoidal dilation have been noted in liver biopsy, though not specific to the condition.[7] Massive hepatic necrosis is commonly seen in ALF induced by HLH. Etiopathogenesis of liver injury can be explained by the rapid permeation of activated hemophagocytic histiocytes or cytokine flooding or due to the progression of the underlying disease.[8] There is an increasing incidence of HLH and its complications being reported in adults in the last decade. The diagnostic dilemma occurs early in the course of the disease as there are no clinical or laboratory features specific to this condition. Hence, HLH should always be suspected in any acute liver failure of unexplained etiology. One of the key features in diagnosing HLH is the demonstration of hemophagocytes in the biopsy of bone marrow or liver. Serial bone marrow and liver biopsies may be helpful in strongly suspected cases.[9] In general, the diagnosis of HLH portends a poor prognosis with a very high mortality rate[5] depending on the underlying disease, age, ferritin level,[10] and so on.
CONCLUSION
HLH should be one of the differentials in any patient presenting with acute liver failure of unexplained etiology. Early diagnosis and timely initiation of specific therapy may improve clinical outcomes.
Acknowledgment
I thank my professors and my colleagues for their guidance and support throughout.
Declaration of patient consent
Patient’s consent is not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Hemophagocytic lymphohistiocytosis: When the immune system runs amok. Klin Padiatr. 2009;221:278-85.
- [CrossRef] [PubMed] [Google Scholar]
- Primary and secondary hemophagocytic lymphohistiocytosis: Clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6:137-54.
- [CrossRef] [PubMed] [Google Scholar]
- Viral etiology, clinical and laboratory features of adult hemophagocytic lymphohistiocytosis. J Med Virol. 2016;88:541-9.
- [CrossRef] [PubMed] [Google Scholar]
- Hemophagocytic lymphohistiocytosis: Clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-5.
- [CrossRef] [PubMed] [Google Scholar]
- Multiorgan failure due to hemophagocytic syndrome: A case report. Cases J. 2008;1:209.
- [CrossRef] [PubMed] [Google Scholar]
- Hepatic manifestations of hemophagocytic syndrome: A study of 30 cases. Am J Gastroenterol. 2001;96:852-7.
- [CrossRef] [PubMed] [Google Scholar]
- Pathology of the liver in familial hemophagocytic lymphohistiocytosis. Am J Surg Pathol. 2010;34:852-67.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160:275-87.
- [CrossRef] [PubMed] [Google Scholar]
- Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56:154-5.
- [CrossRef] [PubMed] [Google Scholar]




