
Translate this page into:
A rare presentation of fulminant subacute sclerosing panencephalitis with acute disseminated encephalomyelitis and meningoencephalitis
-
Received: ,
Accepted: ,
How to cite this article: Kumar R, Pandit S, Bhadoria DP, Gautam S. A rare presentation of fulminant subacute sclerosing panencephalitis with acute disseminated encephalomyelitis and meningoencephalitis. Indian J Med Sci 2021;73:348-51.
Abstract
Subacute sclerosing panencephalitis (SSPE) is a rare form of chronic progressive neurodegenerative disorder, caused by persistent central nervous system (CNS) infection by hypermutated strains of Measles virus. It primarily affects children and early adolescent age group. Since there is widespread involvement of CNS, the primary symptoms may start with behavior and personality changes with intellectual involvement followed with myoclonic jerks, convulsion, and hemi or quadriparesis. In later stage, the patient may experience dementia, stupor, coma, or death. However, initial presentation can be variable and atypical, making the diagnosis very challenging in clinical practice. Herein, we report a case of fulminant SSPE in a 13-year-old adolescent male who presented with the features of acute disseminated encephalomyelitis (ADEM) and meningoencephalitis. The patient presented in altered sensorium with the history of poor communications, jerky movement of limbs, and fever for 10–14 days. Preliminary investigations were suggestive of meningoencephalitis and ADEM. After initial course of treatment, the patient did not improve, deteriorated further, and succumbed to his illness, a day before we received the conclusive reports for SSPE. As we experienced from our case report, SSPE can create a lot of diagnostic dilemma to the clinicians in such a fulminant presentation, mainly due to limitation of resources, time, and lack of proper vaccination records. The lack of definitive treatment for SSPE once again proved the proverb that “prevention will always be better than cure.”
Keywords
Subacute sclerosing panencephalitis
Acute disseminated encephalomyelitis
Electroencephalograph
Cerebrospinal fluid
Measles

INTRODUCTION
Subacute sclerosing panencephalitis (SSPE) is a rare and fatal form of central nervous system (CNS) infection caused by certain defective form of measles virus. After primary infection of measles (generally before 2 years of age), there is a latent period of 6–8 years, to develop the primary symptoms. SSPE is a rare disease nowadays in developed countries, however; in developing countries like India, frequent presentation is not a surprising element in clinical practice, due to discouraging vaccination coverage and poor socioeconomic status.[1] Initial presentation typically involves behavioral abnormality, poor scholastic performance, intellectual deterioration, and myoclonic jerks. In later stage, patients may suffer further clinical deterioration with symptoms of generalized rigidity, acute blindness, stupor, coma, or death. Clinical staging can be summarized as;[2]
Stage 1: Behavioral decline (lethargy, inattention, or temper tantrums) and cognitive decline.
Stage 2: Myoclonic jerks, seizures, and dementia.
Stage 3: Rigidity, extrapyramidal symptoms, and progressive unresponsiveness.
Stage 4: Coma, vegetative state, autonomic instability, and akinetic mutism.
Majority of the patients die within 1–3 years of illness with few exceptions where spontaneous recovery can be achieved. However, in case of fulminant presentation of SSPE, there is no defined course of illness due to rapid disease progression and diagnostic challenges. Atypical features can be summarized as early or very late onset of disease with ataxia, focal deficit, generalized seizures, non-specific electroencephalograph (EEG) pattern, and overlapped features of acute disseminated encephalomyelitis (ADEM) or meningoencephalitis.[3,4]
CASE REPORT
A 13-year-old male child, born of non-consanguineous marriage with normal vaginal delivery and development milestones, who was apparently well 2 weeks back, developed complaints of poor communication with decreased appetite and physical activity along with jerky movement of all four limbs followed by intermittent low-grade fever for 7–10 days. Subsequently, he developed altered sensorium 2 days back with aggressive behavior and irrelevant talking with loss of bladder and bowel control. Fever was on and off in nature, without chills and rigor or diurnal variation, without any history of headache, rashes, vomiting, and bleeding manifestations. Two days before hospitalization, frequency of jerky movements increased from one episode every 30–40 min to one episode every 5–10 min, involving face and limbs, as informed by parents. There was no history of unconsciousness, frothing from mouth, rolling of eyes, tongue bite, and weakness of limbs. There was no history of any toxic substance exposure, dog bite, drug intake, or trauma. There was no history of similar episodes in the past. The patient was unimmunized and there was no history of any childhood illness such as measles, polio, tuberculosis, or chickenpox. The primary treatment was given by local medical practitioner in the form of antipyrectics and antiepileptic drugs (phenytoin), but there was no symptomatic relief for the same. The patient presented at our hospital on day 14 of illness in altered sensorium with loss of bladder and bowel control associated with generalized rigidity, persistent tonic–clonic seizures refractory to anti-epileptics, and loss of passive body movement. The patient was thin built with Glasgow Coma Score 8, febrile with rapid and shallow breathing (respiratory rate 26/min), pulse was 96/min, and blood pressure was 96 mm of Hg in supine position. In CNS examination, (i) higher mental functions were affected severely (speech and sensorium), (ii) persistent tonic–clonic seizures were present. Catatonia with neck rigidity was observed with positive Kernig’s and Brudzinski sign. (iii) Tone was increased in all four limbs and power was graded zero with bilateral withdrawal plantar response and absence of deep tendon reflexes observed with sluggish reactive pupils. Rest of the systemic examination were within normal limits. The provisional diagnosis was made of meningoencephalitis and primary treatment was started for same with antiviral agents (intravenous acyclovir, 15 mg/kg 8 h), antibacterial (intravenous ceftriaxone and vancomycin, 100 mg/kg/day in two divided dose and 15 mg/kg 6 h, respectively), and anti-epileptic drugs (intravenous levetiracetam 500 mg 12 h) with steroids in lower doses (intravenous dexamethasone 0.15 mg/kg 6 h). Initial NCCT HEAD, chest X-ray, electrocardiography, and ultrasound abdomen reports were normal. Total cell counts were increased in hematology reports with neutrophil predominance; however, rest of the blood investigations (biochemistry and hematology and microbiology including hepatitis B and C, HIV, Vitamin B12 level, and thyroid function test) were in normal limits. In cerebrospinal fluid (CSF) studies, total cells were 88 with neutrophilic predominance, sugar was normal with mildly raised protein and CSF adenosine deaminase: −3 IU/L. CSF reports for India ink, Cryptococcus antigen, acid-fast bacilli, and CBNAAT were negative, with no growth in CSF culture in 48 h. By initial reports, tubercular meningoencephalitis was also ruled out. All the tests for workup of fever were done such as dengue, malaria, chikungunya, and enteric fever and found to be negative. EEG was done next day, showed generalized triphasic spikes every 2–3 s, provisionally suggestive of SSPE/ encephalopathy [Figure 1]. Anti-measles antibody titer samples were sent for both CSF and serum study along with other viral serology. MRI brain with whole spine screening was done, showed confluent hyperintensities in bilateral periventricular deep white matter and diffuse involvement of corpus callosum, indicative of ADEM [Figure 2a and b]. Patient’s condition was getting deteriorated, not responding to the treatment for meningoencephalitis and high-dose steroid (intravenous methylprednisolone 30 mg/kg/day) was started in view of ADEM. After 3 days of subsequent treatment, the patient condition worsened further and developed hypotension with persistent hypoxia and Type 1 respiratory failure. Antibiotics coverage was upgraded, the patient was shifted to ICU facility and put-on ventilator support but later he succumbed to his illness on day 5 of hospitalization. Relatives were counseled repeatedly about poor prognosis during course of illness and explained about the need of postmortem brain biopsy later to establish the final diagnosis, but consent could not be taken for the same. CSF for anti-measles antibody and viral serology reports were received after the death, and reliable for the diagnosis of SSPE in view of extremely high-level anti-measles antibody (IgG) titer in both serum and CSF. Rest of the serology reports (cytomegalovirus, herpes virus, and Epstein–Barr virus) were negative. The final diagnosis was “fulminant SSPE stage-IV with ADEM and meningoencephalitis.”
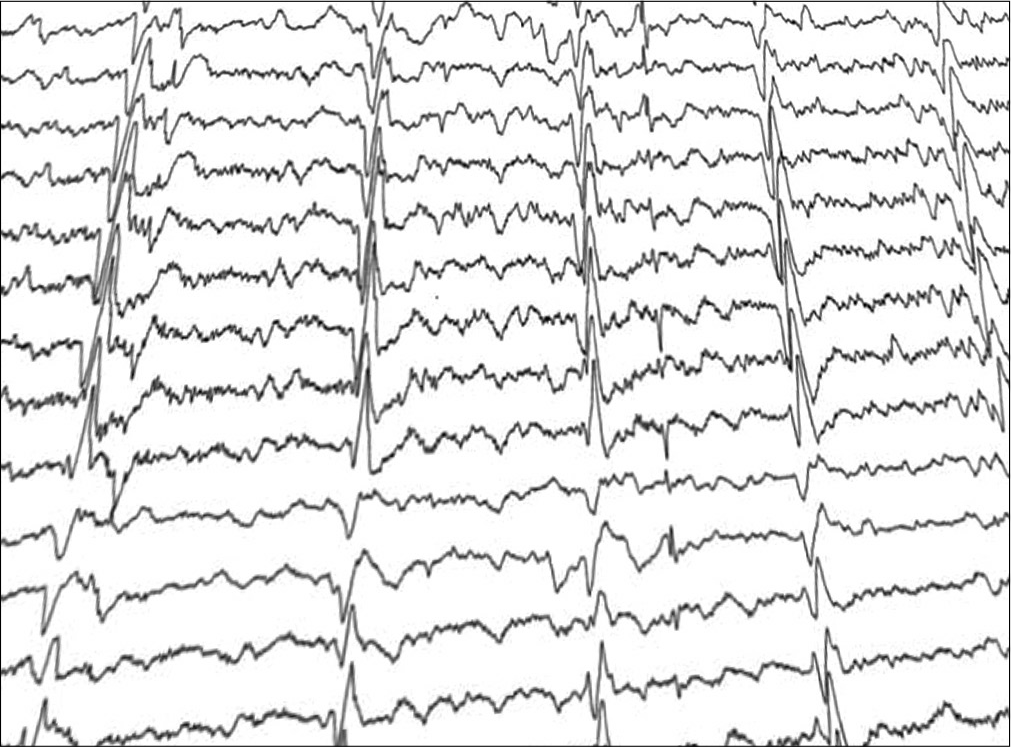
- Generalized triphasic spikes every 2–3 s, suggestive of subacute sclerosing panencephalitis/encephalopathy.
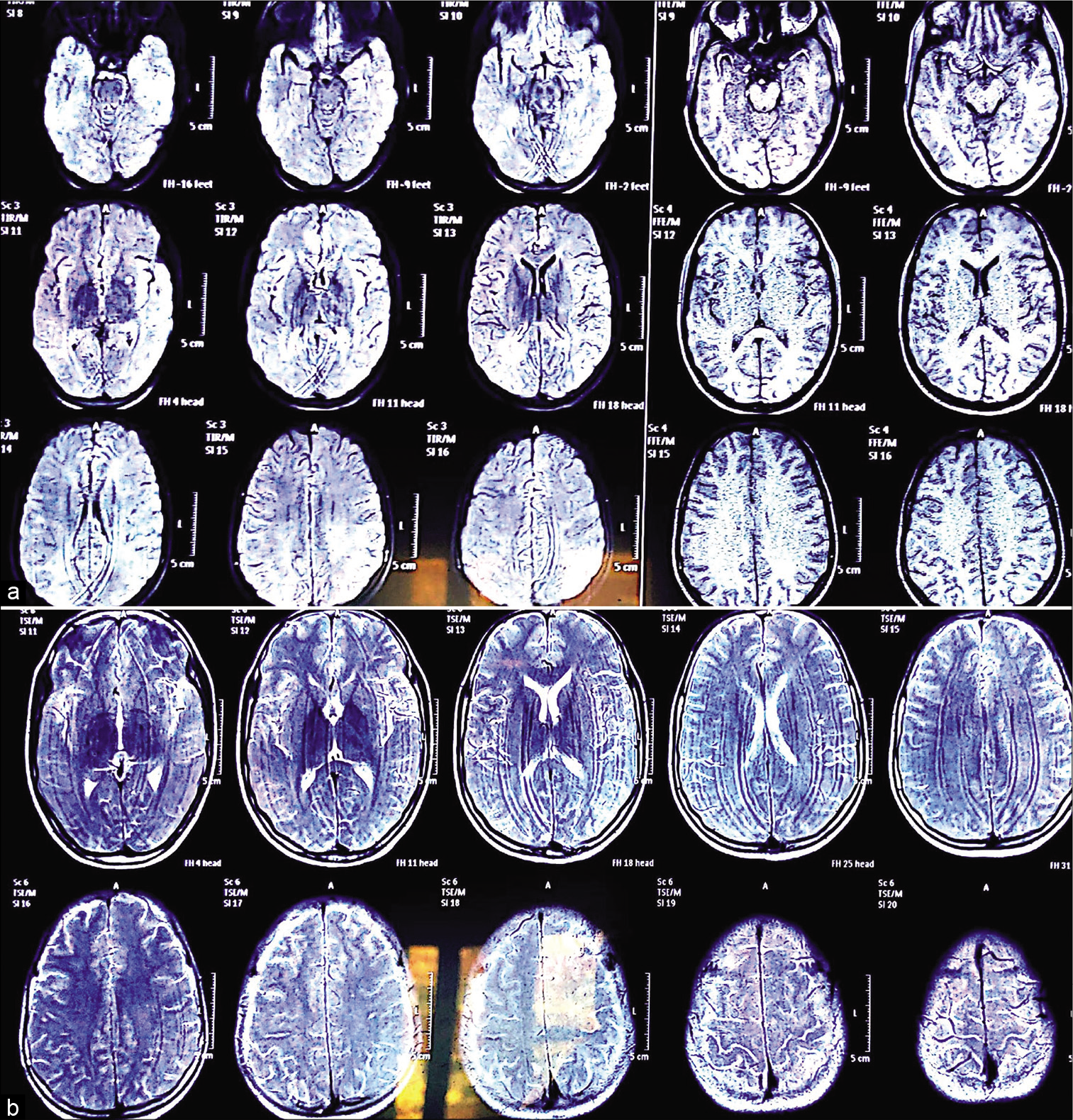
- (a and b) Confluent hyperintensities in bilateral periventricular deep white matter and diffuse involvement of corpus callosum, indicative of acute disseminated encephalomyelitis.
DISCUSSION
SSPE is the most severe sequelae of the measles virus infection with nearly 95% mortality rate, characterized by neurodegenerative changes involves both the gray and white matter, caused by slow acting but defective and hypermutant strains of measles virus.[5] The initial symptoms are very non-specific, affect behavior and intellectual abilities, and can involve motor function in the form of myoclonic jerks and limb weakness. In final stages, patients may experience visual disturbances, autonomic involvement, and finally stupor, coma, or death.[6] In general, SSPE takes about 1–3 years to fully involve the CNS and autonomic nervous system, and the fulminant presentation advances to death in average duration of 6 months. The initial presentation with focal neurological deficit rarely occurs in SSPE and concurrent CNS infections can hasten the progression to death within in few weeks that can mislead the diagnosis, especially in younger age group, toward major causes of acute encephalitis in India such as Japanese encephalitis and enteroviruses. For reliable diagnosis of SSPE, fulfillment of any three of the five criteria required, as proposed by Dyken;[7]
CLINICAL: Progressive, subacute mental deterioration with myoclonus
EEG: Periodic, stereotyped, high voltage discharges
CSF: Raised gamma-globulin or oligoclonal pattern
Measles antibodies: Raised titer in serum (>1:256) and/ or CSF (>1:4)
Brain biopsy: Suggestive of panencephalitis.
There is no adequate therapy available for SSPE in medical science, but if diagnosis can be achieved in earlier stages, certain drugs such as interferon therapy, isoprinosine, and amantadine can prolong life if long-term therapy is given.[8] At present, the only measure that can effectively prevent the incidence of this deadly sins in all age groups is universal vaccination coverage and registration of all suspected and confirmed cases of measles virus by strict surveillance. Measles vaccine can evidently prevent the SSPE incidence not only by its protective efficacy for measles infection but also indirectly through herd immunization, but it requires a larger proportion to be covered by extended vaccination programs.[9] In India, due to its huge population burden and poor socioeconomic status, the incidence is very high in comparison to developed countries and many more could be the submerged part of iceberg due to limited reporting of cases and lack of effective immunization coverage. That’s why, we should always include SSPE as a differential in measles-endemic country like India in such kind of fulminant presentation so that early effective treatment can be started.
CONCLUSION
In short, SSPE is a slowly progressive but fatal CNS disease, affects mainly the patients from backward socioeconomic classes. Since there can be symptoms similar with other CNS diseases, anti-measles antibody titer should be included as routine investigation for such kind of presentation, as it may progress to the fatal state in few weeks after the initial presentation. The treatment available is very costly and too far from the reach of many health centers in country and is also not definitively curative, but can be used to prolong some portion of life. At present, the only solution for effective fight against SSPE is 100% immunization coverage for children and females from child-bearing age. Since situation can be very stressful for family members of such patients with poor socioeconomical status, a dreaded curse-like SSPE may further complicates their financial and emotional status. They require a huge emotional support and empathy from the society and medical fraternity.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Early-onset subacute sclerosing panencephalitis: Report of two cases and review of literature. Ann Indian Acad Neurol. 2019;22:361-3.
- [CrossRef] [PubMed] [Google Scholar]
- Subacute sclerosing panencephalitis: An update. Dev Med Child Neurol. 2010;52:901-7.
- [CrossRef] [PubMed] [Google Scholar]
- Fulminant subacute sclerosing panencephalitis presenting with acute ataxia and hemiparesis in a 15-year-old boy. J Clin Neurol. 2014;10:354-7.
- [CrossRef] [PubMed] [Google Scholar]
- Rapid progressive subacute sclerosing panencephalitis after perinatally acquired measles virus infection. Lancet. 1995;345:1124.
- [CrossRef] [Google Scholar]
- Trial of intraventricular ribavirin therapy for subacute sclerosing panencephalitis in Japan. Brain Dev. 2003;25:514-7.
- [CrossRef] [Google Scholar]
- Subacute sclerosing panencephalitis with atypical onset: clinical, computed tomographic, and magnetic resonance imaging correlations. J Child Neurol. 2000;15:258-60.
- [CrossRef] [PubMed] [Google Scholar]
- Subacute sclerosing panencephalitis. Current status. Neurol Clin. 1985;3:179-96.
- [CrossRef] [Google Scholar]
- Treatment of subacute sclerosing panencephalitis with interferon-alpha, ribavirin, and inosiplex. J Child Neurol. 2002;17:703-5.
- [CrossRef] [PubMed] [Google Scholar]
- Review of the effect of measles vaccination on the epidemiology of SSPE. Int J Epidemiol. 2007;36:1334-48.
- [CrossRef] [PubMed] [Google Scholar]




